
Discovery of two new Cu–Sn chalco–halides for potential solar absorber applications
Journal of Materials Chemistry A Royal Society of Chemistry (RSC) (2026)
Abstract:
New compounds are discovered in the under-explored d 10 –s 2 (Cu–Sn) family using exploratory synthesis guided by computational tools. Band-gaps in the visible region with moderate charge-carrier mobilities make these potential solar absorbers. We explore multiple-cation chalco–halide phase fields evaluated by their synthetic accessibility using machine learning models. Exploratory synthesis guided by computational tools leads to the discovery of two new compounds; CuSn 2 SI 3 and Cu 0.35 Sn 5.29 S 2 I 7 , their structures, and electronic and optical properties are reported herein. This is the first report of a stable quaternary compound in the Cu–Sn–S–I phase field. The two new compounds show related crystal structures where Sn 4 S 2 I 4 layers are a common structural motif in both. These Sn 4 S 2 I 4 layers are connected by Cu 2 I 2 layers and disordered Cu–Sn–I layers, forming the three-dimensional structures of CuSn 2 SI 3 and Cu 0.35 Sn 5.29 S 2 I 7 respectively. Electronic band structure calculations using density functional theory show the presence of a direct band gap in CuSn 2 SI 3 and suggest anisotropic transport, in line with the layered structure of the compound. A mixture of the two compounds with ∼86% CuSn 2 SI 3 , shows a band gap in the visible region, close to 2.1 eV and a significant photo-induced charge carrier mobility of ∼1.3 cm 2 V −1 s −1 . This demonstrates Cu–Sn chalco–halides can form a promising phase space to explore for solar absorber materials, with further design and tuning of band gap.Strong correlations in lanthanide interfaces and Metal Oxides
(2023)
Abstract:
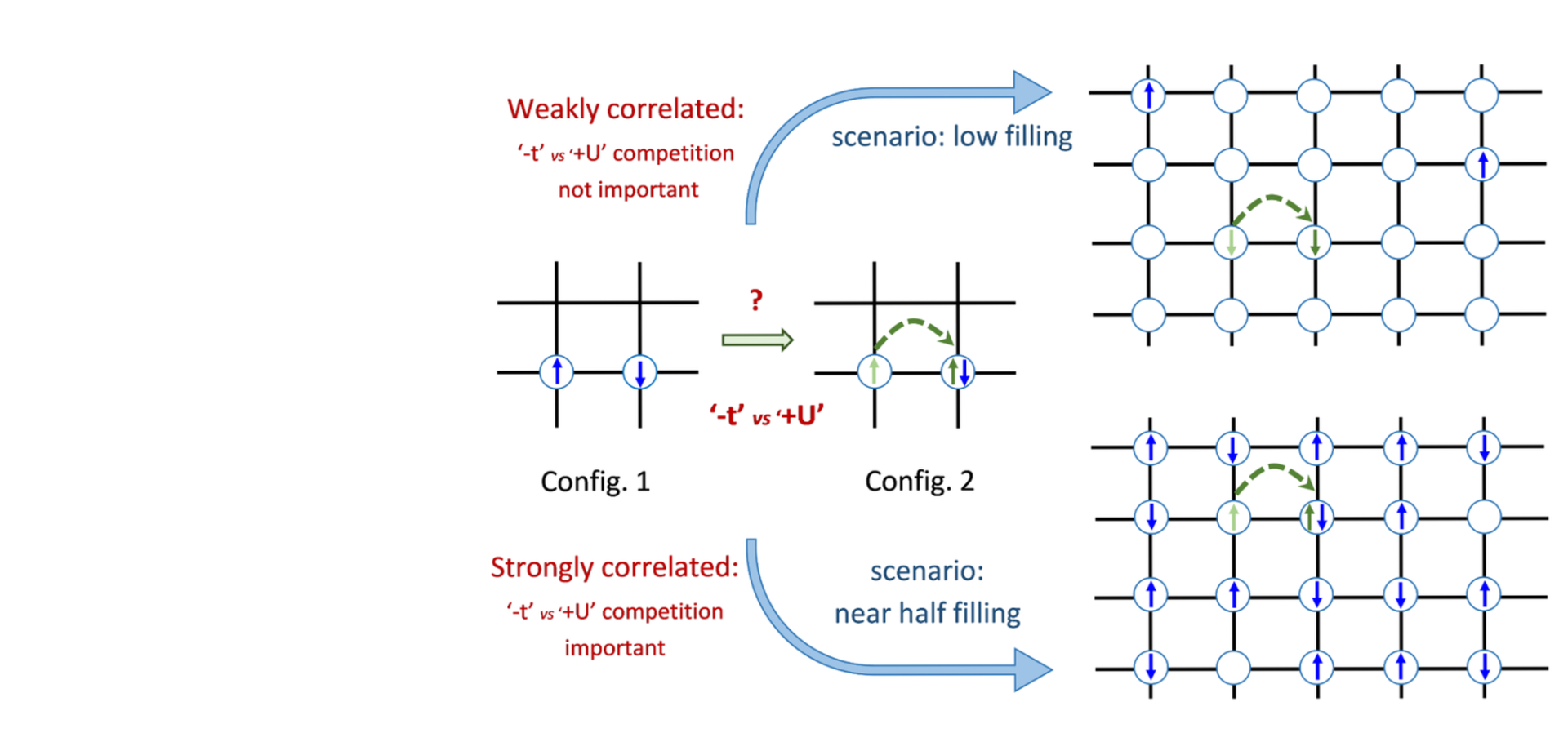
A complete canonical description for why the world and its constituents behave the way they do is a daunting undertaking. Unexpectedly it is so even though the nature and the mathematical form of electronic interactions that account for the energy scales relevant for everyday life, and by that scope the realm of condensed matter physics and material science, have been known for about a century now. The difficulty is that for a collection of large number of many-body entities which can be electrons or atoms, emergent phenomena arise with features that cannot be comprehended by the underlying laws. This makes the contemporary theoretical condensed matter research dealing with electronic structures a scientific exercise in comparison of various levels of approximations. The primary overarching challenge to the theoretical understanding of materials can be attributed to the mathematical and computational complexity that arises in the calculation of the electron-electron interaction term in a realistic set-up. When the effect of this term dominates the behaviour of a material, the material is said to be strongly correlated. One such class of materials are lanthanides and lanthanide metal oxides. Lanthanide compounds are usually associated with a complex phase diagram often involving metal-insulator transitions under a range of conditions viable for industrial applications where mean-field approaches fail. This thesis addresses the realistic modelling of their electronic structure in bulk and as thin films taking into account the effects of interfaces as is needed for understanding the origin of observed behaviours and their applications. We start with a discussion of the various models, approximations, and formalism used throughout this thesis. We further follow it with the study of a model 4-layer correlated interface described by a Hubbard-like Hamiltonian intending to set the premise for realistic calculations. We analyse how the interplay of various kinetic energy terms and strong correlations influence the observation of Mott transition in this model system. We interpret this simplified set-up as a quantum double-well and investigate the impact of correlations and bias on resonant tunnelling conditions. Following this we study Samarium monochalcogenides which show a pressure induced isostructural insulator to metal transition characterised by the presence of an intermediate valence state at higher pressure that cannot be captured by density functional theory. These materials, particularly SmS and SmSe, have been proposed to serve as the piezoresistive component of an alternative form of technology that uses piezoelectric transistors. This transistor operation is based on the compression of the piezoresistive component by the piezoelectric part which undergoes an insulator to metal transition either continuously or discontinuously and therefore can serve as switches or memory elements, respectively. The failure of the mean-field corrections incorporated within density functional theory is ascribed to the presence of strongly correlated Sm-4f orbitals which lead us to the use of dynamical mean field theory corrections. As a direct outcome of including the charge and spin fluctuations incorporated in dynamical mean field theory framework within the scope of the Hubbard-I approximation, we see the emergence of insulating and metallic phases with increasing pressure as a function of changing valence. This is accompanied by significantly improved predictions of the equilibrium lattice constants and bulk moduli for SmS, SmSe, and SmTe verifying experiments. We then perform nudged elastic band analysis that reveals that the insulating states are characterised by a finite quasiparticle weight that decreases as the gap closes with pressure rendering the transition to be not Mott-like. As a result, our work classifies these materials as correlated band insulators. Since we observe the 4f electrons becoming more localised in their high pressure metallic state, we study their behaviour in a piezoresistive element-like set-up where we simulate a few layers of Sm-monochalcogenides in a heterostructure set up with tungsten leads. We find that in the thin film configuration where the transport mechanism becomes tunnelling, the 4f electrons do participate in transmission at and around the Fermi level and thus applying the appropriate level of theory is crucial to the prediction of length scales for their use as piezoresistive elements. We analyse the behaviour of the f-states around the interface, obtain the transmission through the junction at equilibrium and compare transmission ratios at different junction length scales to define an OFF state for the use of SmSe as a piezoresistive element in transistor type operations. We then look at lanthanum metal oxides and the role of correlations and defects in determining their experimentally observed properties. The first set of such materials investigated are two double perovskites La2TiFeO6 and La2VCuO6 which are candidates for promising spintronic applications if the charge ordering between Ti-Fe and V-Cu could be controlled experimentally. However after obtaining these structures with pulsed laser deposition and studying their properties experimentally, no ordered phases could be found and only partial charge transfer could be observed. We first study their ground state properties that show a competition between two different charge transfer type phases are responsible for the observed disorder in La2VCuO6. For La2TiFeO6, we employ random structure search and find that multiple low lying phases with comparable frequencies lead to a high configurational entropy. On studying the entropy forming ability of all 400 optimised structures, we find that the disorder and accompanying partial charge transfer so observed experimentally could be attributed to the high value of configurational entropy which is enhanced by the presence of Lanthanum vacancies to a larger extent and also over oxygenation to a lesser extent. Finally we consider CeO2 and its mixed metal oxide with Zr doping called zirconia to understand and compare the role of oxygen vacancies that leads to their use as heterogeneous catalysts. We compare the qualitative and quantitative results from density functional theory with and without incorporation of a static Coulomb repulsion term to account for the strong electronic correlations in Ce. We compute formation energies for oxygen defects and find that both DFT and DFT+U show Zr adsorbed on CeO2 is energetically favourable for vacancy formation although surprisingly the inclusion of the effect of correlations in the DFT+U framework reduces this favourability. However this is restored on including the effect of lattice dynamics stemming from the observation of soft phonon mediated symmetry breaking that facilitates this vacancy formation along with an increase in effective mass. The research elaborated in this thesis emphasises the role of different levels of state of-the-art theoretical methods in incorporating the effect of strong correlations. We comment on the success of these approaches in determining structural transitions, associated device scaling, and vacancy effects for various ongoing industry relevant applications using lanthanides in bulk, as thin films, and lanthanide metal oxides, respectively. This provides an overview for conceptual understanding of the impact of many-body effects in the design and use of these materials for practical applications.Pressure-induced electronic transitions in samarium monochalcogenides
Physical Review B American Physical Society 105:19 (2022) 195135
Abstract:
The pressure-induced isostructural insulator-to-metal transition for SmS is characterized by the presence of an intermediate valence state at higher pressure which cannot be captured by density functional theory. As a direct outcome of including the charge and spin fluctuations incorporated in dynamical mean-field theory, we see the emergence of insulating and metallic phases with increasing pressure as a function of changing valence. This is accompanied by significantly improved predictions of the equilibrium lattice constants and bulk moduli for all Sm monochalcogenides verifying experiments. Nudged elastic band analysis reveals the insulating states to have a finite quasiparticle weight, decreasing as the gap closes rendering the transition to be not Mott-like, and classifies these materials as correlated band insulators. The difference between the discontinuous and continuous natures of these transitions can be attributed to the closeness of the sharply resonant Sm-4f peaks to the Fermi level in the predicted metallic states in SmS compared with SmSe and SmTe.Study of disorder in pulsed laser deposited double perovskite oxides by first-principle structure prediction
npj Computational Materials Springer Science and Business Media LLC 7:1 (2021) 92