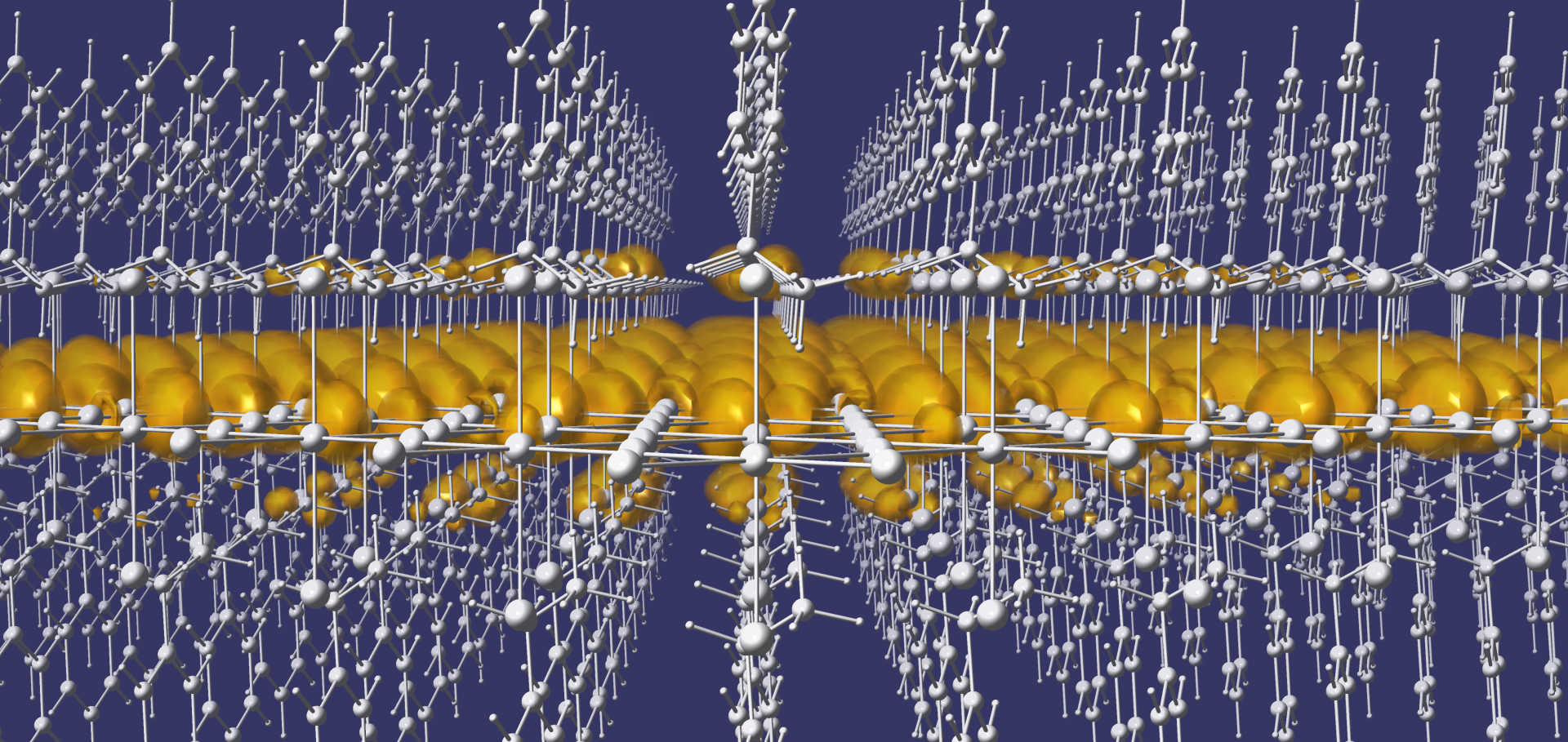

Our research is centered on the study and design of novel semiconductors using first principles computational modeling techniques such as density functional theory (DFT) and many body perturbation theory (MBPT). The premise of first principles calculations is that physical properties of materials can be calculated starting from the basic laws of Quantum Mechanics, without any empirical assumptions, and using only chemical and structural information as input parameters.
We have a wide-ranging expertise in first principles calculations of structural, vibrational, electronic, transport and optical properties of novel semiconductors and insulators, as well as developing efficient strategies for automated high-throughput screening through large sets of chemical compositions. We combine first principles methods with analytical modeling and data science techniques with the goal to uncover materials design rules and accelerate materials discovery by developing new physical and chemical intuition.