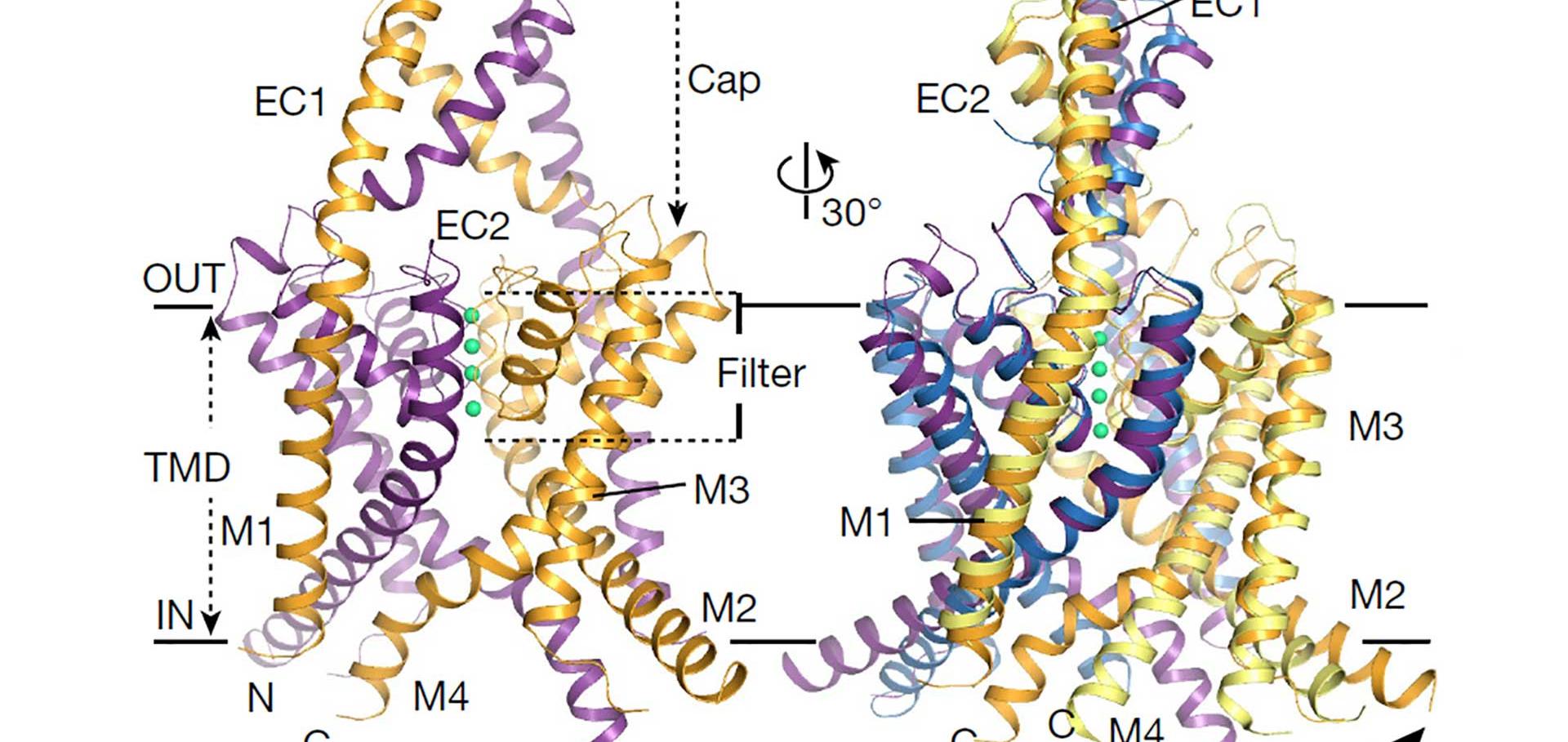
Involvement of the N-terminus of Kir6.2 in the inhibition of the KATP channel by ATP.
J Physiol 514 ( Pt 1):Pt 1 (1999) 19-25
Abstract:
1. ATP-sensitive potassium (KATP) channels are composed of pore-forming Kir6.2 and regulatory SUR subunits. A truncated isoform of Kir6.2, Kir6.2DeltaC26, expresses ATP-sensitive channels in the absence of SUR1, suggesting the ATP-inhibitory site lies on the Kir6. 2 subunit. 2. We examined the effect on the channel ATP sensitivity of mutating the arginine residue at position 50 (R50) in the N-terminus of Kir6.2, by recording macroscopic currents in membrane patches excised from Xenopus oocytes expressing wild-type or mutant Kir6.2DeltaC26. 3. Substitution of R50 by serine, alanine or glycine reduced the Ki for ATP inhibition from 117 microM to 800 microM, 1.1 mM and 3.8 mM, respectively. The single-channel conductance and kinetics were unaffected by any of these mutations. Mutation to glutamate, lysine, asparagine, glutamine or leucine had a smaller effect (Ki, approximately 300-400 microM). The results indicate that the side chain of the arginine residue at position 50 is unlikely to contribute directly to the binding site for ATP, and suggest it may affect ATP inhibition by allosteric interactions. 4. Mutation of the isoleucine residue at position 49 to glycine (I49G) reduced the channel ATP sensitivity, while the mutation of the glutamate residue at position 51 to glycine (E51G) did not. 5. When a mutation in the N-terminus of Kir6.2DeltaC26 that alters ATP sensitivity (R50S; Ki, 800 microM) was combined with one in the C-terminus (E179Q; Ki, 300 microM), the Ki for the apparent ATP sensitivity was increased to 2.8 mM. The Hill coefficient was also increased. This suggests that the N- and C-termini of Kir6.2 may co-operate to influence channel closure by ATP.The N-terminus of Kir6.2 is involved in coupling to SUR1
BIOPHYSICAL JOURNAL 76:1 (1999) A14-A14
PIP2 and PIP as determinants for ATP inhibition of KATP channels.
Science 282:5391 (1998) 1141-1144
Abstract:
Adenosine triphosphate (ATP)-sensitive potassium (KATP) channels couple electrical activity to cellular metabolism through their inhibition by intracellular ATP. ATP inhibition of KATP channels varies among tissues and is affected by the metabolic and regulatory state of individual cells, suggesting involvement of endogenous factors. It is reported here that phosphatidylinositol-4, 5-bisphosphate (PIP2) and phosphatidylinositol-4-phosphate (PIP) controlled ATP inhibition of cloned KATP channels (Kir6.2 and SUR1). These phospholipids acted on the Kir6.2 subunit and shifted ATP sensitivity by several orders of magnitude. Receptor-mediated activation of phospholipase C resulted in inhibition of KATP-mediated currents. These results represent a mechanism for control of excitability through phospholipids.Mechanism of ATP-sensitive K channel inhibition by sulfhydryl modification.
J Gen Physiol 112:3 (1998) 325-332
Abstract:
ATP-sensitive potassium (KATP) channels are reversibly inhibited by intracellular ATP. Agents that interact with sulfhydryl moieties produce an irreversible inhibition of KATP channel activity when applied to the intracellular membrane surface. ATP appears to protect against this effect, suggesting that the cysteine residue with which thiol reagents interact may either lie within the ATP-binding site or be inaccessible when the channel is closed. We have examined the interaction of the membrane-impermeant thiol-reactive agent p-chloromercuriphenylsulphonate (pCMPS) with the cloned beta cell KATP channel. This channel comprises the pore-forming Kir6.2 and regulatory SUR1 subunits. We show that the cysteine residue involved in channel inhibition by pCMPS resides on the Kir6.2 subunit and is located at position 42, which lies within the NH2 terminus of the protein. Although ATP protects against the effects of pCMPS, the ATP sensitivity of the KATP channel was unchanged by mutation of C42 to either valine (V) or alanine (A), suggesting that ATP does not interact directly with this residue. These results are consistent with the idea that C42 is inaccessible to the intracellular solution, and thereby protected from interaction with pCMPS when the channel is closed by ATP. We also observed that the C42A mutation does not affect the ability of SUR1 to endow Kir6.2 with diazoxide sensitivity, and reduces, but does not prevent, the effects of MgADP and tolbutamide, which are mediated via SUR1. The Kir6.2-C42A (or V) mutant channel may provide a suitable background for cysteine-scanning mutagenesis studies.Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP.
J Gen Physiol 112:3 (1998) 333-349