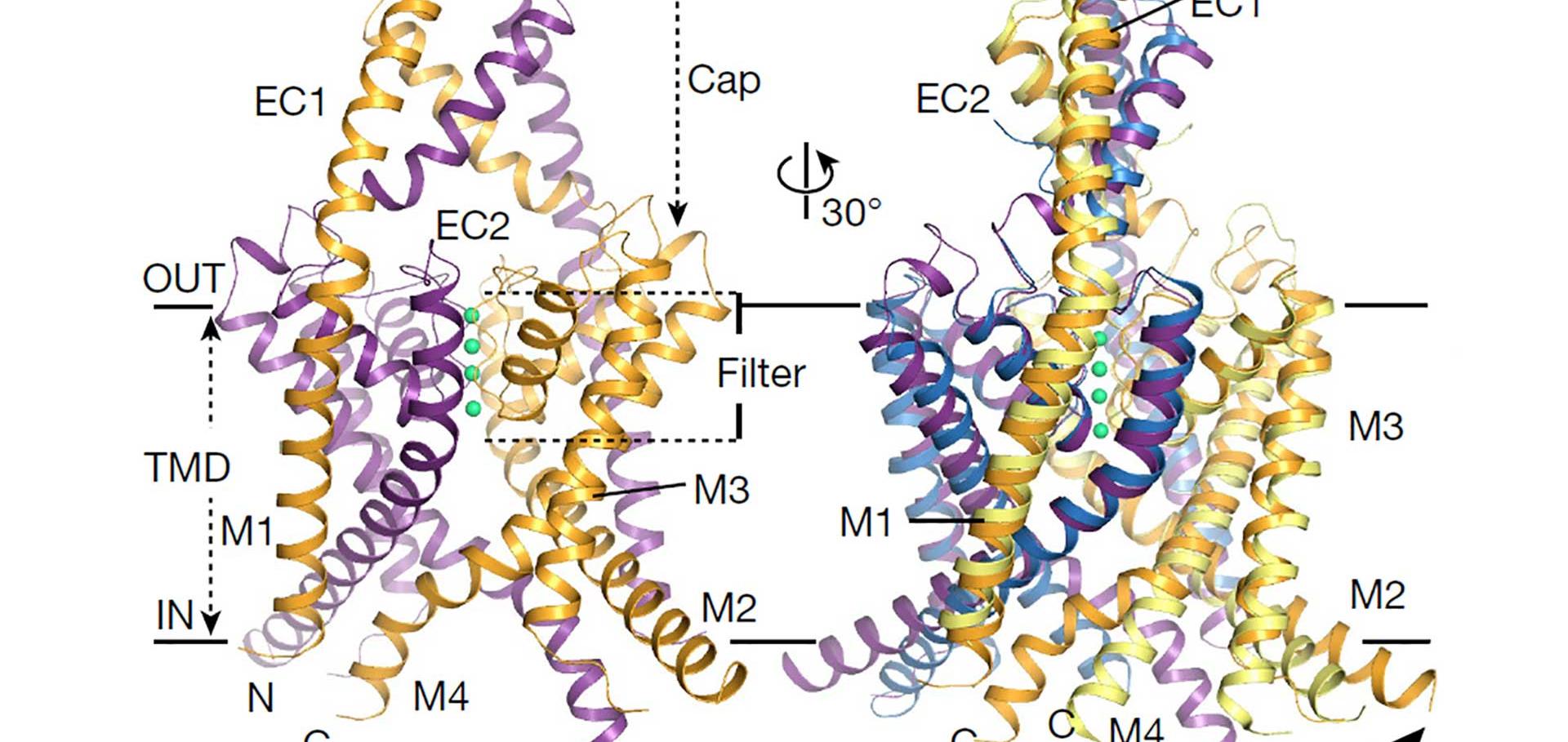
A conserved drug-binding site controls the selectivity filter gate in K2P K+ channels
ACTA PHYSIOLOGICA 219 (2017) 78-78
Regulation of Two-pore Domain K plus Channels by Natural Effectors and Pharmacological Agents
ACTA PHYSIOLOGICA 221 (2017) 64-64
The structural movement of the TM4 segment during pore gating in TREK1 channels
ACTA PHYSIOLOGICA 219 (2017) 80-80
Functional annotation of ion channel structures by molecular simulation
Structure Cell Press 24:12 (2016) 2207-2216
Abstract:
Ion channels play key roles in cell membranes, and recent advances are yielding an increasing number of structures. However, their functional relevance is often unclear and better tools are required for their functional annotation. In sub-nanometer pores such as ion channels, hydrophobic gating has been shown to promote dewetting to produce a functionally closed (i.e. non-conductive) state. Using the serotonin receptor (5-HT3R) structure as an example, we demonstrate the use of molecular dynamics to aid the functional annotation of channel structures via simulation of the behavior of water within the pore. Three increasingly complex simulation analyses are described: water equilibrium densities; single ion free energy profiles; and computational electrophysiology. All 3 approaches correctly predict the 5-HT3R crystal structure to represent a functionally closed (i.e. non-conductive) state. We also illustrate application of water equilibrium density simulations to annotate to different conformational states of a glycine receptor.Correction: Dominant-Negative Effect of a Missense Variant in the TASK-2 (KCNK5) K+ Channel Associated with Balkan Endemic Nephropathy
PLOS ONE Public Library of Science (PLoS) 11:7 (2016) e0160114