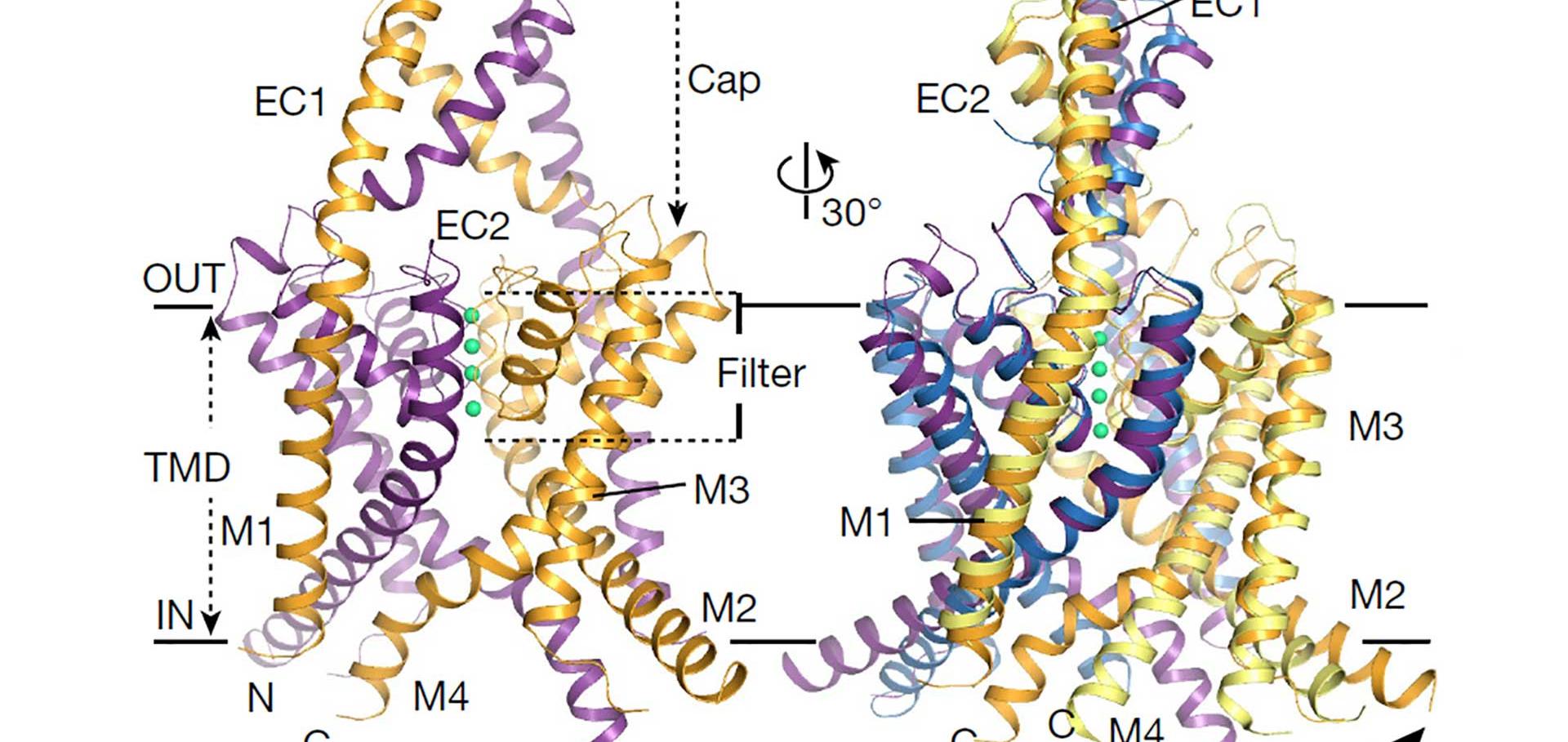
H-Bonding at the Helix-Bundle Crossing Controls Gating in Kir Potassium Channels
Neuron 55 (2007) 602-614
Molecular dynamics simulations of inwardly rectifying (Kir) potassium channels: a comparative study.
Biochemistry 46:12 (2007) 3643-3652
Abstract:
Inward rectifier potassium (Kir) channels regulate cell excitability and transport K+ ions across membranes. Homotetrameric models of three mammalian Kir channels (Kir1.1, Kir3.1, and Kir6.2) have been generated, using the KirBac3.1 transmembrane and rat Kir3.1 intracellular domain structures as templates. All three models have been explored by 10 ns molecular dynamics simulations in phospholipid bilayers. Analysis of the initial structures revealed conservation of potential lipid interaction residues (Trp/Tyr and Arg/Lys side chains near the lipid headgroup-water interfaces). Examination of the intracellular domains revealed key structural differences between Kir1.1 and Kir6.2 which may explain the difference in channel inhibition by ATP. The behavior of all three models in the MD simulations revealed that they have conformational stability similar to that seen for comparable simulations of, for example, structures derived from cryoelectron microscopy data. Local distortions of the selectivity filter were seen during the simulations, as observed in previous simulations of KirBac and in simulations and structures of KcsA. These may be related to filter gating of the channel. The intracellular hydrophobic gate does not undergo any substantial changes during the simulations and thus remains functionally closed. Analysis of lipid-protein interactions of the Kir models emphasizes the key role of the M0 (or "slide") helix which lies approximately parallel to the bilayer-water interface and forms a link between the transmembrane and intracellular domains of the channel.Conformational rearrangements during PIP2 gating in Kir channels
BIOPHYS J (2007) 27A-28A
Identification of gain of function mutations in KirBac potassium channels by genetic complementation in K+ auxotrophic strains of E-coli and yeast.
BIOPHYS J (2007) 104A-104A
Control of pH and PIP2 gating in heteromeric Kir4.1/Kir5.1 channels by H-Bonding at the helix-bundle crossing.
Channels (Austin) 1:5 (2007) 327-330