Engineering III-V nanowires for optoelectronics: from epitaxy to terahertz photonics
Proceedings of SPIE Society of Photo-optical Instrumentation Engineers 10543 (2018)
Abstract:
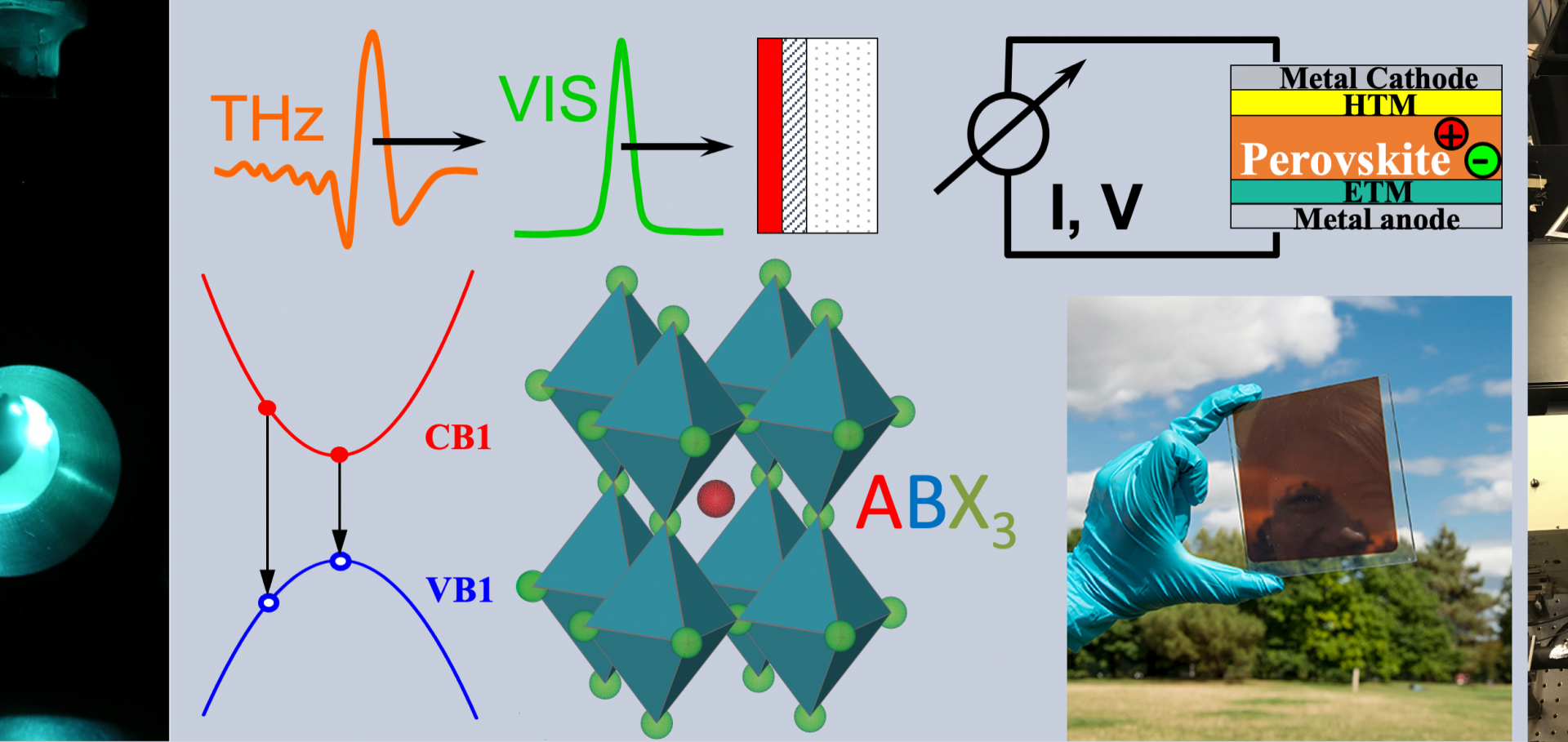
Downloading of the abstract is permitted for personal use only. Nanowires show unique promise as nanoscale building blocks for a multitude of optoelectronic devices, ranging from solar cells to terahertz photonic devices. We will discuss the epitaxial growth of these nanowires in novel geometries and crystallographic phases, and the use of terahertz conductivity spectroscopy to guide the development of nanowire-based devices. As an example, we will focus on the development of nanowire-based polarization modulators for terahertz communications systems.Bimolecular recombination in methylammonium lead triiodide perovskite is an inverse absorption process
Nature Communications Springer Nature 9 (2018) 293
Abstract:
Photovoltaic devices based on metal halide perovskites are rapidly improving in efficiency. Once the Shockley–Queisser limit is reached, charge-carrier extraction will be limited only by radiative bimolecular recombination of electrons with holes. Yet, this fundamental process, and its link with material stoichiometry, is still poorly understood. Here we show that bimolecular charge-carrier recombination in methylammonium lead triiodide perovskite can be fully explained as the inverse process of absorption. By correctly accounting for contributions to the absorption from excitons and electron-hole continuum states, we are able to utilise the van Roosbroeck–Shockley relation to determine bimolecular recombination rate constants from absorption spectra. We show that the sharpening of photon, electron and hole distribution functions significantly enhances bimolecular charge recombination as the temperature is lowered, mirroring trends in transient spectroscopy. Our findings provide vital understanding of band-to-band recombination processes in this hybrid perovskite, which comprise direct, fully radiative transitions between thermalized electrons and holes.The Route to Nanoscale Terahertz Technology: Nanowire-based Terahertz Detectors and Terahertz Modulators
Institute of Electrical and Electronics Engineers (IEEE) 00 (2018) 1-2
Photocurrent spectroscopy of perovskite solar cells over a wide temperature range from 15 to 350 K
Journal of Physical Chemistry Letters American Chemical Society 2018:9 (2017) 263-268
Abstract:
Solar cells based on metal halide perovskite thin films show great promise for energy generation in a range of environments from terrestrial installations to space applications. Here we assess the device characteristics of the prototypical perovskite solar cells based on methylammonium lead triiodide (CH3NH3PbI3) over a broad temperature range from 15 to 350 K (−258 to 77 °C). For these devices, we observe a peak in the short-circuit current density and open-circuit voltage at 200 K (−73 °C) with decent operation maintained up to 350 K. We identify the clear signature of crystalline PbI2 contributing directly to the low-temperature photocurrent spectra, showing that PbI2 plays an active role (beyond passivation) in CH3NH3PbI3 solar cells. Finally we observe a blue-shift in the photocurrent spectrum with respect to the absorption spectrum at low temperature (15 K), allowing us to extract a lower limit on the exciton binding energy of 9.1 meV for CH3NH3PbI3.Band Tail States in FAPbI3: Characterization and Simulation
Fundacio Scito (2017)