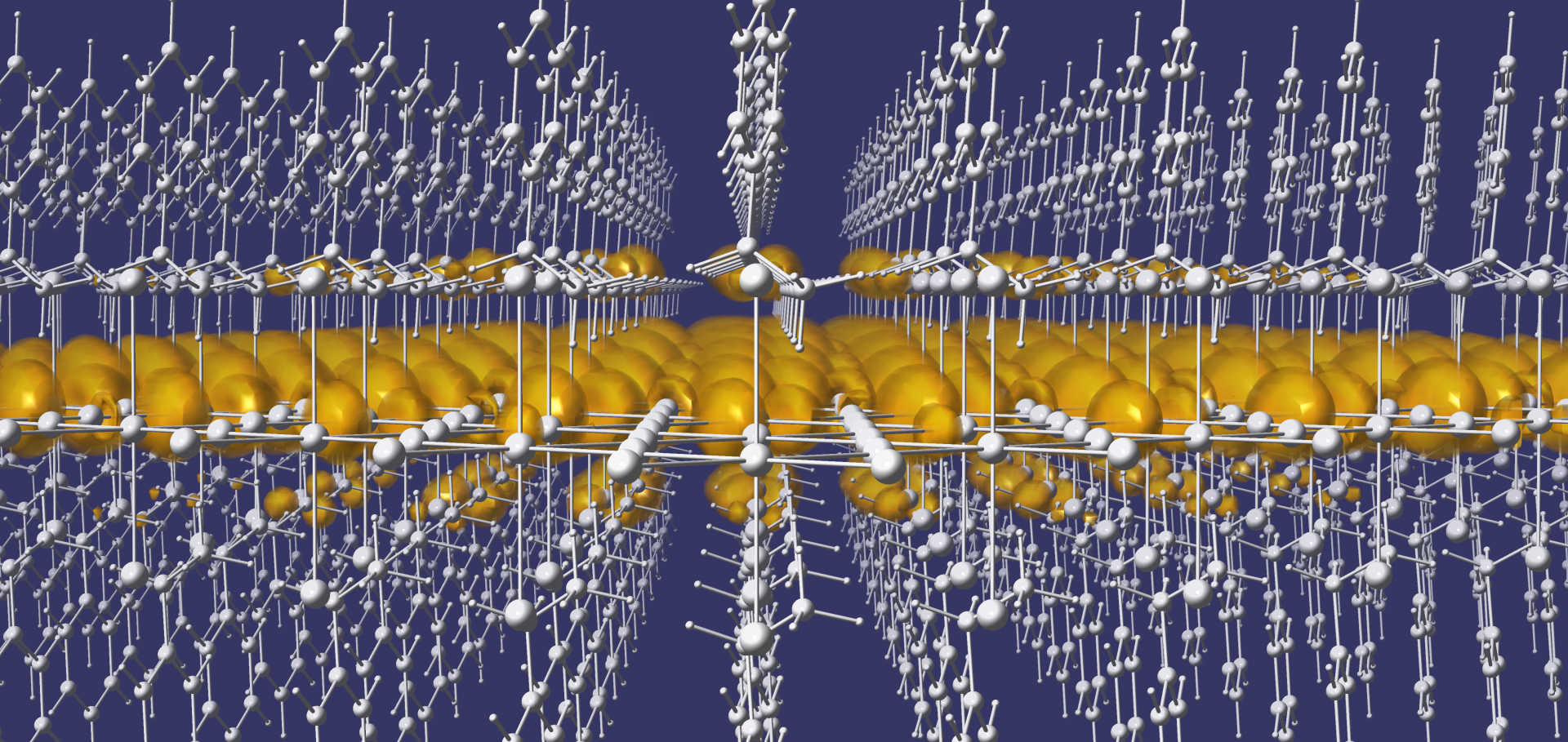
Addition to “Tuning the Quantum-Well Structure of Single-Crystal Layered Perovskite Heterostructures”
Twisted Tin‐Chloride Perovskite Single‐Crystal Heterostructures
Abstract:
Self‐assembly affords simpler synthetic routes to heterostructures compared with manual layer‐by‐layer stacking, yet controlling interlayer twist angles in a bulk solid remains an outstanding challenge. We report two new single‐crystal heterostructures: (Sn2Cl2)(CYS)2SnCl4 (CYS = +NH3(CH2)2S–; Sn_CYS) and (Sn2Cl2)(SeCYS)2SnCl4 (SeCYS = +NH3(CH2)2Se–; Sn_SeCYS) synthesized in solution, with alternating perovskite and intergrowth layers. Notably, compared to the recently reported lead analog, (Pb2Cl2)(CYS)2PbCl4 (Pb_CYS), the tin heterostructures feature a twist between the perovskite and intergrowth layers. We trace this twist to local distortions at the Sn centers, which change the interfacial lattice‐matching requirements compared to those of the Pb analog. Electronic band structure calculations show that the striking differences in the relative energies of perovskite‐ and intergrowth‐derived bands in Sn_CYS and Pb_CYS arise from structural and not compositional differences. The structural anisotropy of Sn_CYS is also reflected in a large in‐plane photoluminescence linear anisotropy ratio. Interfacial strain further affords differential incorporation of Pb into the perovskite and intergrowth layers of the Sn heterostructures, resulting in redshifted optical absorption onsets. Thus, we posit that local structural distortions may be exploited to manipulate the twist angle and interfacial strain in bulk heterostructures, providing a new handle for tuning the band alignments of bulk quantum‐well electronic structures. Replacing lead with tin in a single‐crystal halide perovskite heterostructure drives a twist between the perovskite (gray) and intergrowth (blue) layers. The accompanying structural distortions and interfacial strain change the calculated orbital composition of the band edges and enable high in‐plane optical anisotropy in the Sn analog. (VBT = valence band top; CBB = conduction band bottom).Discovery of high-temperature charge order and time-reversal symmetry-breaking in the kagome superconductor YRu3Si2.
Abstract:
Identifying high-temperature unconventional charge order and superconductivity in kagome systems is crucial for understanding frustrated, correlated electrons and enabling future quantum technologies. Here, we report that the kagome superconductor YRu3Si2 hosts an exceptional interplay of charge order, magnetism, and superconductivity, revealed through a comprehensive suite of muon spin rotation (μSR), magnetotransport, X-ray diffraction, and density functional theory (DFT). We identify a high-temperature charge-ordered state with propagation vector (1/2,0,0) and a record onset temperature of 800 K, unprecedented in kagome systems and quantum materials more broadly. μSR measurements further reveal time-reversal symmetry-breaking below 25 K and field-induced magnetism near 90 K, features mirrored in the magnetoresistance, which reaches 45% at low temperatures. Band-structure calculations show two van Hove singularities near the Fermi level, including one within a flat band. At low temperatures, YRu3Si2 becomes superconducting below Tc = 3.4 K with either two full isotropic gaps or an anisotropic nodeless gap. These results establish YRu3Si2 as a prime platform for studying correlated kagome physics.Tailoring a lead-free organic–inorganic halobismuthate for large piezoelectric effect
Abstract:
Molecular piezoelectrics are a potentially disruptive technology, enabling a new generation of self-powered electronics that are flexible, high performing, and inherently low in toxicity. Although significant efforts have been made toward understanding their structural design by targeted manipulation of phase transition behavior, the resulting achievable piezoresponse has remained limited. In this work, we use a low-symmetry, zero-dimensional (0D) inorganic framework alongside a carefully selected ‘quasi-spherical’ organic cation to manipulate organic–inorganic interactions and thus form the hybrid, piezoelectric material [(CH3)3NCH2I]3Bi2I9. Using variable–temperature single crystal X-ray diffraction and solid-state nuclear magnetic resonance spectroscopy, we demonstrate that this material simultaneously exhibits an order–disorder and displacive symmetry-breaking phase transition. This phase transition is mediated by halogen bonding between the organic and inorganic frameworks and results in a large piezoelectric response, d 33 = 161.5 pm/V. This value represents a 4-fold improvement on previously reported halobismuthate piezoelectrics and is comparable to those of commercial inorganic piezoelectrics, thus offering a new pathway toward low-cost, low-toxicity mechanical energy harvesting and actuating devices.Tuning the quantum-well structure of single-crystal layered perovskite heterostructures
Abstract:
Single-crystal layered perovskite heterostructures provide a scalable platform for potentially realizing emergent properties recently seen in mechanically stacked monolayers. We report two new layered perovskite heterostructures M2(PbCl2)(AMCHC)2(PbCl4)·2H2O (1_M where M = Na+, Li+; AMCHC = +NH3CH2C6H10COO–) crystallizing in the chiral, polar space group C2. The heterostructures exhibit alternating layers of a lead-chloride perovskite and an intergrowth comprising corner-sharing PbCl4(η2-COO)2 polyhedra with bridging equatorial chlorides and terminal axial oxygen ligands. Small alkali metal cations and water molecules occupy the cavities between the polyhedra in the intergrowth layer. The heterostructures display wide bandgaps, two closely spaced excitonic features in their optical spectra, and strong second harmonic generation. The calculated band structure of 1_Na features a Type-I quantum-well structure, where the electron–hole correlation function corresponding to the lowest excited state points to electron–hole pairs localized within a single inorganic layer (intralayer excitons), as seen in typical layered halide perovskites. In contrast, calculations show that 1_Li adopts a Type-II quantum-well structure, with electrons and holes in the lowest excited state residing in different inorganic layers (interlayer excitons). Calculations on model complexes suggest that these changes in band alignment, between Type-I and Type-II quantum-well structures, are driven by the placement of the alkali metal and the orientation of the water molecules, changing the electrostatic potential-energy profiles of the heterostructures. Thus, this study sets the stage for accessing different alignments of the perovskite and intergrowth bands in bulk perovskite heterostructures that self-assemble in solution.