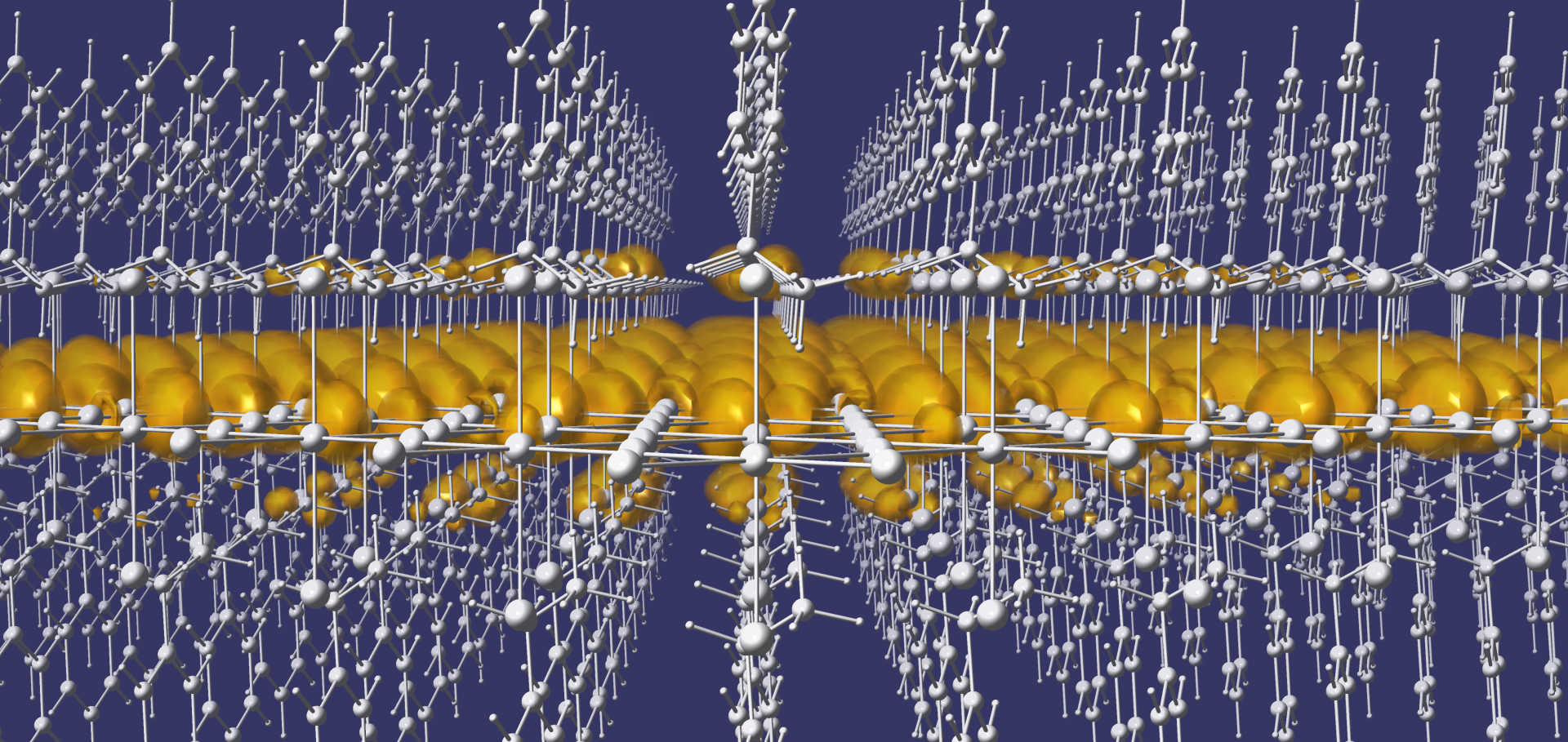
Anharmonic phonons in the high-temperature phase of KNiCl<sub>3</sub>.
Structural dynamics (Melville, N.Y.) 12:5 (2025) 054103
Abstract:
The high-temperature phase of the hexagonal halide perovskite KNiCl3 is investigated using time-of-flight single crystal neutron diffraction at 633 K (360 °C). Phonons are captured through thermal diffuse scattering, integrated in energy but resolved in momentum. Harmonic phonon calculations based on density functional theory yield imaginary phonon frequencies for this phase, indicating the presence of structural instabilities at this level of theory. It is shown that the inclusion of anharmonic phonon-phonon interactions removes these instabilities, leading to good qualitative agreement with the experimental diffuse scattering. These results demonstrate that the high-temperature phase of KNiCl3 is stabilized by anharmonic phonon-phonon interactions.Ruddlesden–Popper Defects Act as a Free Surface: Role in Formation and Photophysical Properties of CsPbI 3
Advanced Materials Wiley (2025) 2501788
Abstract:
The perovskite semiconductor, CsPbI3, holds excellent promise for solar cell applications due to its suitable bandgap. However, achieving phase‐stable CsPbI3 solar cells with high power conversion efficiency remains a major challenge. Ruddlesden–Popper (RP) defects have been identified in a range of perovskite semiconductors, including CsPbI3. However, there is limited understanding as to why they form or their impact on stability and photophysical properties. Here, the prevalence of RP defects is increased with increased Cs‐excess in vapor‐deposited CsPbI3 thin films while superior structural stability but inferior photophysical properties are observed. Significantly, using electron microscopy, it is found that the atomic positions at the planar defect are comparable to those of a free surface, revealing their role in phase stabilization. Density functional theory (DFT) calculations reveal the RP planes are electronically benign, however, antisites observed at RP turning points are likely to be malign. Therefore it is proposed that increasing RP planes while reducing RP turning points offers a breakthrough for improving both phase stability and photophysical performance. The formation mechanism revealed here can apply more generally to RP structures in other perovskite systems.Ultrafast Spontaneous Exciton Dissociation via Phonon Emission in BiVO$_4$
(2025)
Ruddlesden-Popper defects act as a free surface: role in formation and photophysical properties of CsPbI3
(2025)
Theory of ab initio downfolding with arbitrary range electron-phonon coupling
(2025)